




核酸の電気泳動

電気泳動とは・・・
核酸(DNAやRNA)やタンパク質の分子サイズや電荷の差を利用し、電場中の電気泳動用ゲルの分子ふるい効果を利用して分離する手法です。

電気泳動では、アガロースやポリアクリルアミドを担体とする「ゲル」を使用します。比較的分子の大きい核酸はアガロースゲルを用いることが多く、酵素処理した断片などはポリアクリルアミドゲルを使用することがあります。タンパク質の分離にはポリアクリルアミドゲルを用い、ゲルの性状、バッファー組成など様々な組み合わせで多種多様な電気泳動方法が存在します。
ここでは、アガロースゲルを用い、核酸(主にDNA)を分離する手法について紹介します。
(アトー社 アガロースゲル電気泳動と染色・蛍光検出撮影のコツ より)
■電気泳動の流れ






ゲルの作製
アガロース粉末を泳動バッファーと混和し、加熱溶解、ゲル化させます。60℃以下に冷ましたゲルをゲルトレイに流し込み、ゲルが固まるのを待ちます。
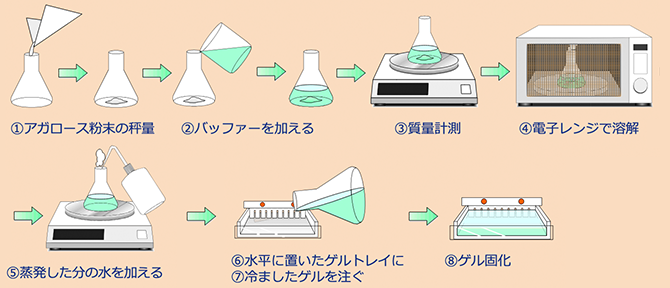
- ①アガロースゲルの濃度を決め、秤量します*1。
バッファー量:ゲルの幅(W)×長さ(L)×厚み(H)cm=AmL
アガロース量:A×ゲル濃度%=Bg
ゲル厚は3~5mmが広く使用されています。ゲル厚は薄い方がバンドがシャープに見えますが、物理的強度は弱くなるので気をつけてください。 - ②TAE*2またはTBE*3バッファーをメスフラスコでAmL計量し、アガロースの粉末と混ぜます。
- ③アガロース、バッファーを三角フラスコに入れたら全体の質量を量り、記録しておきます。
- ④電子レンジまたは湯せんなどでアガロースを完全に溶解します。電子レンジを使用する場合は突沸*4 に注意してください。沸騰してきたら停止させ、泡立てないよう攪拌し、再度あたためます。
- ⑤アガロースが完全に溶けたら*5、三角フラスコを秤に載せ、質量を量ります。加熱により蒸発した水分は蒸留水を追加し、記録しておいた質量に合わせ、ゲル濃度を保ちます。ゲル溶液は約60℃以下になるまで冷ましておきます*7。
- ⑥水準器を用いて、ゲルトレイを水平に設置します*6。微妙な調整は、ゲルトレイの下に紙などを敷いて行ってください。
- ⑦コウムをセットしたゲルトレイにゲル溶液を注ぎます*7。
- ⑧しばらく放置しゲルが固まるのを待ちます。
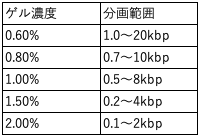
*1:アガロースの種類や濃度は、分離したい核酸の分子量により決めてください。概ね0.5~3%が用いられます。
*2:TAE:40mMトリス、40mM氷酢酸、1mMEDTA
*3:TBE:89mMトリス、89mMホウ酸、2mMEDTA
*4:特にゲル濃度が高い場合(2%以上)注意が必要。加熱中は電子レンジから離れないようにしてください。
*5:溶液中のきらきらした粉末が見えなくなります。溶解が不十分だとゲルが均一に固まらず、泳動パターンが乱れる要因になります。
*6:ゲルトレイが傾いているとゲル厚が不均一になり、泳動パターンの異常要因になることがあります。
*7:ゲルトレイに使用されているアクリルなどの樹脂は、熱により変形の恐れがあります。60℃以下に冷ましてからゲル溶液を流し込みます。
* メーカーにより耐熱性ゲルトレイの提供がある場合はこの限りではありません。
【関連試薬・消耗品】
・アガロース粉末
・泳動バッファー
・ゲルトレイ
試料調製・分子量マーカーの選択
ローディングバッファーの調製を行い、試料をローディングバッファーで希釈調製します。分子量マーカーは分子量の推定だけでなく、電気泳動自体の良否の確認も行えます。
- ●ローディングバッファーの調製
TEバッファーにBPB(ブロムフェノールブルー:紺)、XC(キシレンシアノール:空色)等のマーカー色素を入れ、ショ糖で比重を付け、ローディングバッファーとします。マーカー色素は0.001~0.01%程度、薄く色がつく位の濃度にします*8。
▼希釈時のバンドイメージ

*8:試薬グレードや保存劣化により泳動パターンが乱れることがあります。「WSE-7040 EzApply DNA」は泳動先端に色素マーカーがあり、バンドがシャープになる比重液を採用しています。マーカー色素が多いと泳動パターンの乱れや計測時のバックグラウンドの不均一などの原因になります。特にXCはバンドと重なることが多ので注意ください。
- ●試料溶液の調製
試料をローディングバッファーで希釈調製します。適度に希釈してアプライすることで、バンドがシャープになります。目安としては1バンドあたり10~100ng程度がよいでしょう*9。レーンごとのアプライ量が同じになるように希釈・調製することを推奨します。 - ●試料は要時調製してください。希釈すると保存中にDNA・RNAの分解・吸着など正確な結果が得られない場合があります。
- ●分子量マーカーを用意
分子量の推定だけでなく、電気泳動自体の良否の確認も行えるので、一緒に泳動することをお勧めします。*9:試料の濃度・絶対量が多くなるとバンドが太くなっていきます。
電気泳動
泳動用バッファーとして、ゲルを作製した同じバッファーを泳動槽に入れ、試料をゲルに注入し、電気泳動します。
試料添加
- ●全てのウェルに試料溶液を添加すること、添加容量を同じにすることを推奨します。各レーンへの電荷をそろえることでパターンが乱れることを防ぎます。試料数がレーン数に満たない場合は、ローディングバッファーを空きレーンに添加します。
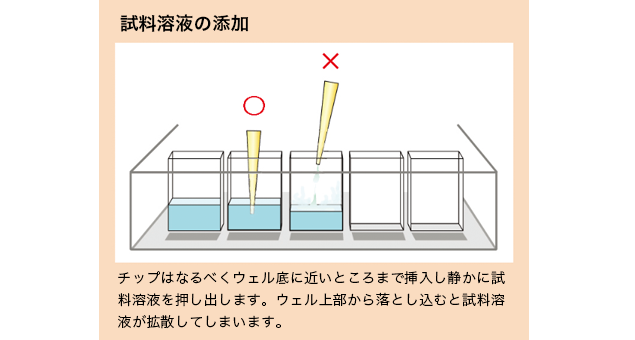
- ●基本的にマーカーは両側に添加します。レーン数が多いときは中心レーンにも流しておくと後で分子量の計測精度が向上します。
- ●ウェルに試料溶液を添加する場合には、チップをウェルの上方から真直ぐ降ろすようにします。チップの挿入位置、サンプル液の注入スピード(ゆっくり静かに)、最後の液の押し出しの有無など条件を同じにしてください。
通電
- ●アガロースゲル電気泳動装置
アガロースゲルの電気泳動に使用されるサブマリンタイプの電気泳動槽は基本構造がシンプルです。なので、試料・ゲル・バッファー等がプロトコル通り調製され、泳動装置のマニュアル通りに使用することで、きれいな泳動パターンが得られます。 - ①泳動用バッファーとして、ゲルの作製で使用したバッファー(TAEやTBEなど)を用意します。泳動槽にゲルが1〜3mm沈む程度の量を入れます。多過ぎるとバンドのシャープさが無くなったり、泳動時間が長くなります。
- ②ウェルに試料と分子量マーカーを添加します。
- ③通電条件を電源部・装置に設定し通電を開始します。試料添加から通電までは時間をおかずに実施します。基本的には電圧一定で泳動します。外部から電源装置をつなげて使用する泳動槽の場合は、電極間距離(陽極と陰極の白金線の間の距離)に合わせ、5~10V/cmを標準にして定電圧条件で泳動してください。電極間距離20cmの場合は100~200V定電圧が標準設定です。
- ④色素が泳動先端または予定の位置まできたら通電を停止します。または、ルーチン的に電気泳動を実施する場合は、泳動条件(バッファー・ゲル・通電条件)が同じため、タイマーを使用して泳動時間を同じにすると再現性が高い実験が可能です。
ゲル染色
核酸の検出は蛍光色素を用いることがほとんどです。一般的な有色色素も使用可能ですが、感度が低い為使用頻度は少ないです。蛍光色素染色は主に安価で実績のあるエチジウムブロマイド(EtBr)が利用されています。1本鎖の核酸や要求感度が高い場合はSYBR Green/Goldなどを使用します。最近ではより安全性の高い蛍光色素が利用されます。
- ①蛍光染色試薬は発ガン性・変異原性などがある為、操作時は必ずグローブを着用してください*10。
- ②染色溶液を濃度ムラの無いように調製します*11。
例:EtBr染色液は、0.5ug/mL以下が標準的濃度です。調製後、遮光保存してください。 - ③染色用トレイ(ガラス不可)を準備して染色溶液を入れ、泳動後のゲルを浸漬させます。染色時間は色素やゲル厚・大きさによって異なります。
EtBrは概ね10~30分で染色が可能です。SYBR Greenは30~60分、EzStainシリーズは10~30分で染色可能です。染色中はアルミホイル等で遮光することを推奨します。 - ④バックグラウンドが低くなるまで30~60分脱色します。蛍光染色色素により、脱色が不要なものもあります。染色液、脱色した液は施設・自治体の指示に従って処理してください。
*10:蛍光染色試薬の安全性
EtBrなどの蛍光染色試薬は核酸に特異的に結合することから発ガン性・変異原性などが報告されています。操作時は必ずグローブを着用してください。使用後は所定の破棄手順を経て処理してください。
「WSE-7130 EzFluoroStainDNA」「WSE-7135 EzPreStainDNA&RNA」は高感度で安全性の高い染色試薬です。
*11:染色液の調製・保存性
「EzFluoroStainDNA」「EzPreStainDNA&RNA」は冷凍庫から出して完全に透明な橙色、溶解した状態で調製してください。EtBr染色液(希釈したもの)の保存は遮光・室温で1間以内に留めてください。「EzFluoroStainDNA」「EzPreStainDNA&RNA」の染色液は遮光・冷蔵で1間以内なるべく早めに使用してください。SYBR Green/Gold等、概ね保存が不可で、使い捨て(要時調製)が基本です。
【関連試薬・消耗品】
・染色溶液
・染色用トレイ(ガラス不可)
蛍光検出/撮影/解析
蛍光検出について
- ●蛍光の原理(蛍光検出の基本概念)
EtBrに代表される核酸の蛍光染色では、紫外線照射により蛍光物質が励起され蛍光を発します。EtBrなどの蛍光物質は核酸に特異的に結合し、その結合量は核酸の分子量、濃度に依存しています。つまり、分子量が大きく、量が多いバンドはより強く光り、反対に分子量が小さく、量が少ないバンドは蛍光が弱くなります。 - ●励起光源
蛍光物質を励起させる最適な光の波長は物質によって異なります。EtBr、SYBR Green/Goldでは250~370nmの紫外線を使用します。EzFluoroStain DNAシリーズはUVもしくはシアン・青色LED440~530nmで励起して、500~600nmの蛍光により観察できます。
*EzFluoroStainDNAピークEx:250/370/482nm、Em:509nm
EzPreStainDNA&RNAピークEx:270/497nm、Em:522nm
紫外線照射装置を用いる場合、EtBrやSYBR Greenなどの蛍光物質では254nmの短波長が最も検出感度が高くなりますが、核酸が分解されやすくなります。人体にも有害ですので直接見たり浴びないように注意が必要です。フィルターを介したり紫外線プロテクターを着用して目視してください。 - ●撮影用フィルター
検出時に余計な光をカットする為に種々の光学フィルターを用います。
蛍光検出・撮影装置 構造とフィルターの役割
- ●蛍光ゲルの検出及びデータ取得には蛍光検出・撮影装置を使用します。励起光源の紫外線照射装置やLED照射装置と高感度カメラ、その間に適正なフィルターを通して蛍光物質=核酸を検出します。撮影データは撮影装置に付属するプリンタで印刷したり、PCやUSBメモリに保存します。
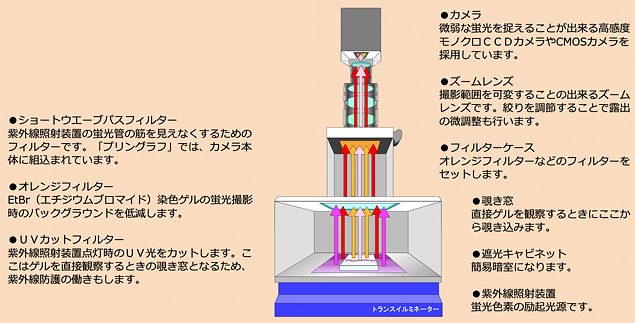
- ●蛍光色素染色をしたゲルは、ラップや紫外線透過型のアクリルトレイに乗せて紫外線・LED照射装置の上に置きます。暗室にして照射を開始し、適切な条件にて検出・撮影・データ保存・印刷をします。
- ●クローニング用のDNAを回収することを目的とする場合、蛍光検出をUVで行うよりもLEDで行うほうがDNAのUVによる分解を妨げるため回収量を多く確保することが可能です。
UV検出の場合も、波長が短くなるほどDNAの分解時間が短くなるのでなるべく長波長のUVを使用することが推奨されます。
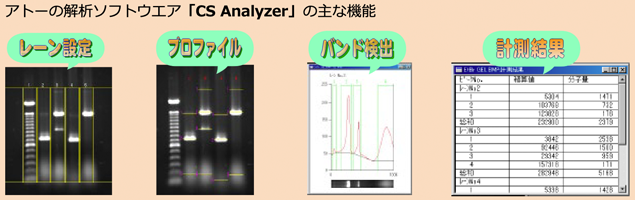
解析
- ●確実なパターン解析を行うために
画像解析ソフトウエアは便利な機能が付いていますが、定量データを出す場合には再現性が求められます。データの再現性を上げる最も良い方法はきれいな電気泳動パターンを得ることです。ここまでに記述してきた内容を参考にして、きれいな電気泳動パターンが得られる実験を行ってください。 - ●濃度定量
蛍光色素染色ゲルのバンドからは、濃度に比例した蛍光強度が得られます。この蛍光強度からバンドの濃度を数値化することが可能です。電気泳動パターン解析ソフトウエアはバンドの濃度を数値化するためのソフトウエアです。濃度を数値化できれば、バンドの濃さを比較したり、濃度推定を行うことが可能です。 - ●分子量の推定
分子量マーカーの各バンドの分子量と移動度を基にして検量線を作成すれば、目的バンドの分子量を推定することが可能です。
核酸抽出/精製
クローニングなどに使用するときは、核酸をゲルから回収して抽出・精製する必要があります。DNAの抽出方法はいくつかあります。ここでは市販のキット(ゲル切り出し精製キットであるQiagen社のQIAquick Gel Extraction Kit)を用いた簡易的な方法を紹介いたします。
QIAquick Gel Extraction Kitを用いたアガロースゲルからのDNA精製
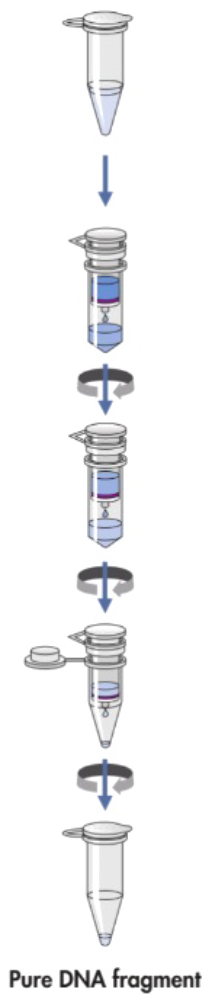
- ①清潔で刃の鋭いメスを用いてDNAフラグメントを含むアガロースゲル部分を切り取る。
- ②無色のチューブにゲルスライスを入れ重量を測定する。ゲル(100mgあるいは約100µL)に3倍容量のBuffer QGを添加する。
- ③50℃で10分間(あるいはゲルが完全に溶解するまで)インキュベートする。 ゲルの溶解をよくするため、インキュベーション中、2~3分毎にチューブをボルテックスして溶液を混和する。
- ④ゲルスライスが完全に溶解後、溶液の色が黄色であることを確認する(アガロース溶解前のBuffer QGの色と同様)。
- ⑤ゲルと等量のイソプロパノールをサンプル溶液に添加し、チューブを数回転倒混和する。
- ⑥2mLのコレクションチューブ(添付)にQIAquickスピンカラムをセットする。
- ⑦DNAを結合させるために、サンプルをQIAquickカラムにアプライして1分間遠心操作する。
- ⑧ろ液を棄て、QIAquickカラムを同じコレクションチューブに戻す。
- ⑨推奨:500µLのBuffer QGをQIAquickカラムに添加し、1分間遠心操作する。
- ⑩洗浄のため750µLのBuffer PEをQIAquickカラムに添加し、1分間遠心する。
- ⑪ろ液を除き、QIAquickカラムをさらに17,900xg(13,000rpm)で1分間遠心操作する。
- ⑫QIAquicカラムを新しい1.5mLのマイクロ遠心チューブにセットする。
- ⑬DNAの溶出を行なうために、50µLのBuffer EB(10mM Tris·Cl、pH8.5)あるいは水(pH7.0~8.5)をQIAquickメンブレン表面の中央に添加し、1分間遠心 する。あるいはDNA濃度を高めるために、30µLの溶出バッファーをQIAquickメンブレンの中央に添加し、1分間カラムを放置後、1分間遠心する。
- ⑭精製したDNAをゲルで解析する際には、Loading Dyeに5倍容量の精製DNAを添加する。ゲルにアプライする前に数回ピペッティングし、よく混和する。
【関連試薬・消耗品】
・ゲル抽出キット
・イソプロパノール
・エタノール(96~100%)
・3M酢酸ナトリウム(pH5.0)




